
Decoding ALS
TAU discovery sheds new light on elusive workings of fatal disease

Researchers from Tel Aviv University have identified a previously unknown process taking place within cells in patients with Amyotrophic Lateral Sclerosis (ALS) that may contribute to the development of treatments for the deadly disease. With ALS, the brain loses its ability to control the body’s muscles. The progressive loss of muscle action in ALS can make it impossible to eat, speak, move and ultimately, to breathe. The disease is typically fatal within two-to-five years of symptom onset. The devastating disease is currently affecting an estimated 450,000 individuals worldwide.
“This discovery provides new encouragement for developing ALS treatments," said the study’s lead researcher, TAU Prof. Eran Perlson from the Department of Physiology and Pharmacology, Sackler Faculty of Medicine and Sagol School of Neuroscience. "It may open new channels for research on the long-term communication between the various nerve regions that are critical for better understanding the pathological processes of ALS." The researchers note, however, that the prospect of treatment development based on the discovery is still years away.
Discovering 'Death Signals'
Experts have made significant advances in ALS research, but its exact cause remains largely unknown and there is currently no effective cure or treatment for the disease. To pave the way for drug development, researchers need better understanding of the basic mechanisms by which ALS attacks and destroys nerve cells in the brain and the spinal cord that control body movement.
The new TAU-led discovery reveals a mechanism in which mutated CRMP4 proteins associated with ALS promote motor neuron loss. The newly-identified mechanism works by initiating a process in which the CRMP4 protein binds dynein, a motor protein that, like a train engine, can take proteins from one side of a neuron to the other. Typically, neurons effectively send signals for muscle movement along their respective nerve fibers (axon), emanating from the neuron cell body (soma) at the spinal cord outward toward the neuron tip at the muscles. This process is essential to maintaining healthy neurons. In ALS, however, toxic ‘death signals’ via CRMP4 and dynein move along the nerve tip to the spinal cord and cause motor neuron death.
The researchers suggest that blocking this mechanism - by genetic or pharmacological means - may reduce symptoms of ALS.
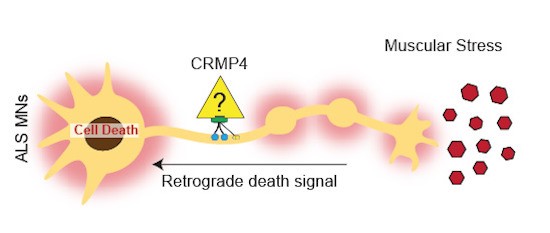
An illustration of the TAU-led discovery of the molecular mechanism that causes nerve cell death in ALS
New Encouragement for ALS Treatments
The researchers used a special biological chip system they developed that allowed them to study how the motor neurons and the musculoskeletal system are destroyed in ALS. Via the unique system, the researchers grew and sorted stem cells into nerve cells. Using advanced biochemical and molecular microscopy methods, the researchers studied the process of nerve cell death in ALS.
The research was led by TAU's Dr. Roy Maimon and postgraduate student Lior Ancol from Prof. Perlson's lab, together with research associate Tal Gardus Pery, Topaz Altman, and Ariel Ionescu. Participating researchers in Israel and abroad included: Prof. Martin Balastik of the Czech Academy of Sciences, Prof. Sami Baramada of the University of Michigan, Dr. Amir Dori of Sheba Medical Center at Tel Hashomer and Prof. Yarden Opatowsky of Bar-Ilan University. The findings of the study were published in the prDeestigious peer-reviewed The EMBO Journal.
Related Articles


